日本発の物質材料研究の
オープンアクセスジャーナル
STAM

英文論文誌 Science and Technology of Advanced Materials(STAM)は、幅広い物質材料研究の各分野で活躍する国内外の専門家39名の編集委員によって編集しています。日本が特に強い物質材料研究の成果を国内外に発信する原著論文誌(ジャーナル)として、日本で唯一の専門研究機関である物質・材料研究機構(NIMS)が刊行を支援し、特徴ある日本の物質材料研究を積極的に企画特集しています。




Citation metrics
- 5.5 (2022) Impact Factor
- Q2 Impact Factor Best Quartile
- 7.3 (2022) 5 year IFQ2 Impact Factor Best Quartile

Speed / acceptance
- 23 days avg. from acceptance to online publication
- 24% acceptance rate
新着論文

新着お知らせ
-
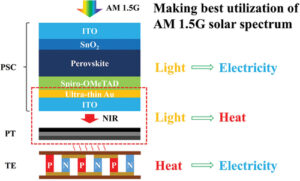 A perovskite solar cell-photothermal-thermoelectric tandem system for enhanced solar energy utilization
A perovskite solar cell-photothermal-thermoelectric tandem system for enhanced solar energy utilization -
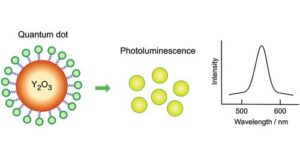 Fabrication of quantum dot-immobilized Y2O3 microspheres with effective photoluminescence for cancer radioembolization therapy
Fabrication of quantum dot-immobilized Y2O3 microspheres with effective photoluminescence for cancer radioembolization therapy -
 Adaptive plasticity of auxetic Kirigami hydrogel fabricated from anisotropic swelling of cellulose nanofiber film
Adaptive plasticity of auxetic Kirigami hydrogel fabricated from anisotropic swelling of cellulose nanofiber film -
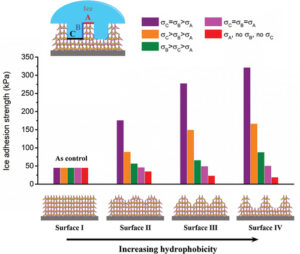 De-icing performance evolution with increasing hydrophobicity by regulating surface topography
De-icing performance evolution with increasing hydrophobicity by regulating surface topography -
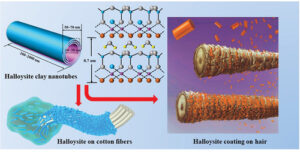 Micropatterning of biologically derived surfaces with functional clay nanotubes
Micropatterning of biologically derived surfaces with functional clay nanotubes -
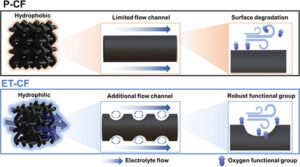 Two-in-one strategy for optimizing chemical and structural properties of carbon felt electrodes for vanadium redox flow batteries
Two-in-one strategy for optimizing chemical and structural properties of carbon felt electrodes for vanadium redox flow batteries -
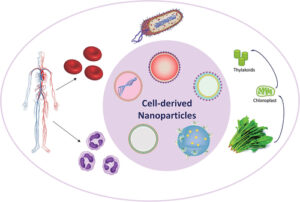 Cell-derived nanomaterials for biomedical applications
Cell-derived nanomaterials for biomedical applications
STAM Methods
STAM 姉妹ジャーナル創刊!

STAMの姉妹誌として2021年に創刊。マテリアルズインフォマティクス、機械学習、ハイスループット合成・評価など、物質材料研究を加速する手法に特化した英文学術論文誌です。STAMと同様にゴールドオープンアクセスジャーナルとして、Web上で誰もが論文を閲覧できます。
ご投稿をお待ちしております!